MATERIALS AND METHODS |
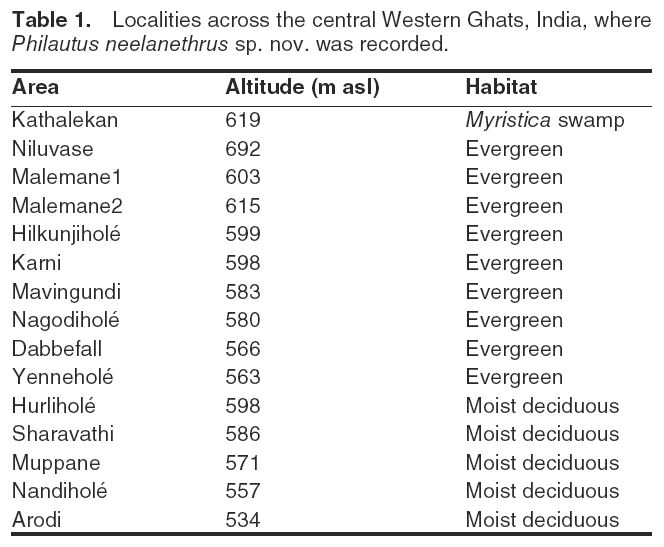
The Sharavathi River basin is situated in the central part of the Western Ghats (Fig. 1). The Sharavathi River originates at Ambuthirtha and flows towards west for about 132 km before joining the Arabian Sea at Honnavar. The type specimens of the new frog species described in this study were collected during stratified systematic sampling (Heyer et al., 1994) with time-constrained and search-all methods (Vasudevan et al., 2001). The type locality is Arodi, Sagar Taluk, Shimoga District, Karnataka state (14°0825N, 74°4744E), 534 m asl (meters above sea level), a moist deciduous forest patch. The region has an undulating terrain, with forests of evergreen and moist-deciduous types. Relatively flat areas within this terrain form a freshwater habitat known as Myristica swamps, dominated by members of plant family Myristicaceae. Localities across the study area where the new species was found are listed in Table 1.
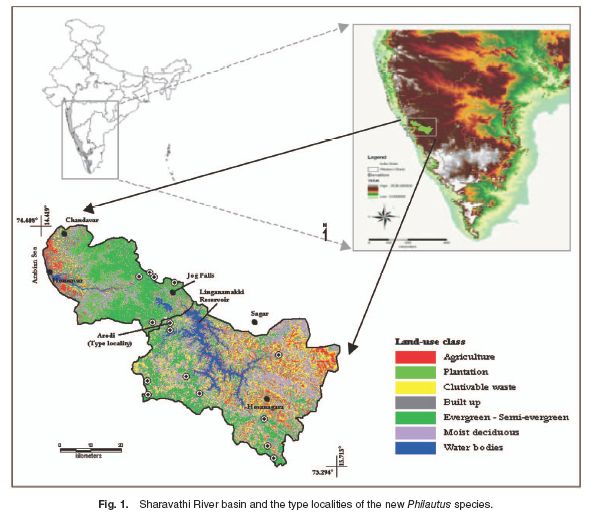
The new frog species was recorded in the study area (Fig. 1) over a period of 4 years since 2001. Although more than 150 specimens were enumerated during multiple field explorations, only nine individuals of the new species (including holotype and paratypes)were collected, on different dates by KVG, NAA and SA. The specimens were used for detailed morphometric description, as well as for molecular analysis to resolve its taxonomic status. Adult specimens of the new frog species were deposited in the Bombay Natural History Museum (BNHS), Mumbai (Holotype, BNHS-4510; Paratype, BNHS-4511) and in the museum of the Zoological Survey of India (ZSI), Kolkata, India (Paratype ZSI-A9866). Specimens were collected from the type locality, photographed, euthanized, and preserved in salt saturated 20% DMSO (dimethyl sulfoxide) solution and/or 80% ethanol. The preserved specimens were used for morphometric studies, and soft tissues taken from the same specimens were used to extract genomic DNA for molecular analysis.
Holotype (BNHS-4510) : adult male, SVL 29.9 mm, collected at Arodi in the Sharavathi River basin on 7 July 2005 by KVG. Paratypes, two adult males: SVL 23.4 mm (ZSI-A9866) collected at Niluvase (13°4418N, 75°0630E; 692 m asl) on 6 November 2003 by KVG; SVL 28.7 mm (BNHS-4511) collected at Arodi (14°0825N,74°4744E; 534 m asl) on 7 July 2005 by KVG.
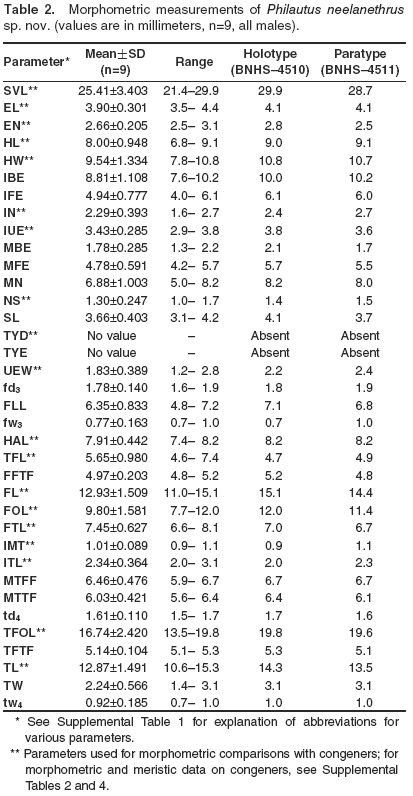
Nine individuals of new species were used for morphometric measurements. Thirty-five morphometric measurements were taken to the nearest 0.1 mm with digital slide calipers (Mitutoyo Corporation,Japan, CD-6BS) for each of the specimens (Table 2). Terminology (Supplemental Table 1) used in the description is based on Bossuyt and Dubois (2001).
A cluster analysis based on unweighted pair-group averages (UPGA) was used to understand the relationship of the new species to other, known congeners (Supplemental Tables 2, 3), and included data on 19 morphometric and three meristic characters (Table 2, Supplemental Table 4). The analysis was carried out using the software package STATISTICA (StatSoft Inc.).
Advertisement calls of the new species were recorded with a digital voice recorder (W-10, Olympus, Japan). A total of 16 calls from seven individuals were recorded at five different localities in the study area. Spectral features of the advertisement calls were analyzed using Sigview (version 1.91) acoustical software (SignalLab,Goran Obradovic).
Call-pattern characteristics of P. neelanethrus sp. nov. (n=16) were also compared with those of one of its closest congeners, P. luteolus, using Bartletts test for homogeneity and significance of variances (Snedecor and Cochran, 1989). The data for P. luteolus, comprising six acoustic parameters based on 51 call samples, were taken from Kuramoto and Joshy (2001), where P. luteolus was referred to as P. cf. travancoricus. Parameters considered were total call duration, call duration in the fast and slow phases, number of pulses in the fast and slow phases, and frequency range.
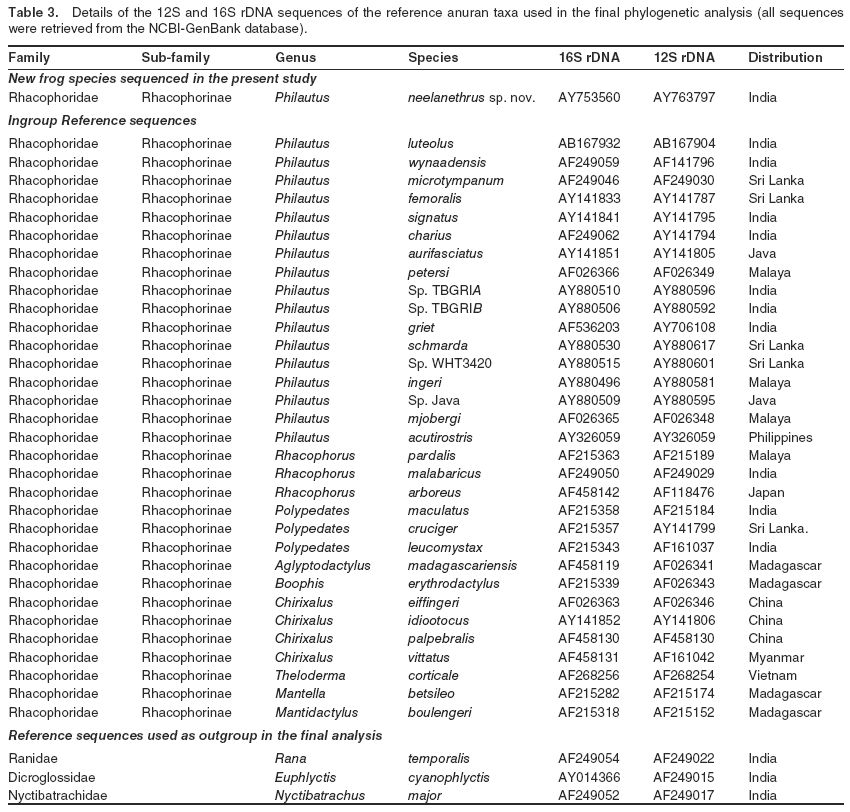
Ribosomal typing was carried out to establish the species status of the new frog taxon. Total genomic DNA was extracted from muscle tissues, taken from preserved specimens collected from different localities, by the proteinase K, phenol-chloroform-isoamyl alcohol method (Shanker et al., 2004). The DNA samples were used to determine molecular diversity across the 12S and 16S mitochondrial rRNA genes, in order to ascertain the species uniqueness and phylogenetic position of the new taxon. Parts of the 16S (~575 bp) and 12S rDNA (~435 bp) genes were amplified and sequenced as described by Dutta et al. (2004). Each sample was sequenced three times for both strands to confirm the sequence data. Sequences have been deposited in GenBank under accession numbers AY763797 (12S rDNA, 415 bp) and AY753560 (16S rDNA, 546 bp).
The 12S and 16S rDNA sequences of the new frog taxon were used in similarity-based BLAST searches of the NCBI-GenBank database (National Center for Biotechnology Information, USA; http://www.ncbi.nlm.nih.gov) to identify related reference anuran species. Initially, corresponding sequences of >100 different reference anuran species were retrieved from the database. The final phylogenetic analysis included reference sequences for only 35 taxa, mainly of different Philautus species (Table 3), to ascertain the phylogenetic position of the new species, and also for possible molecular dating.
All sequences were aligned using the CLUSTAL-X program and then checked for large gaps. The aligned sequences were terminated flush at the ends to avoid missing data for any of the compared reference entries. Three aligned sequence sets, one each for 12S and 16S rDNA and one for combined 12S+16S, were used separately to derive corrected Kimura two-parameter distance estimates (Kimura, 1980) and to infer the phylogenetic position of the new taxon. Neighbor-joining trees were constructed with analytical routines available in the software packages PHYLIP 3.6 (http://evolution.genetics.washington.edu/phylip.html) and MEGA 2.1 (http://www.megasoftware.net). Character state-based maximum likelihood (ML) and maximum parsimony (MP) phylogenetic trees were also constructed using PhyloWin http://pbil.univ-lyon1.fr/software/phylowin.html). In order to test for earliest branching patterns, all substitutions were considered, and separate analyses were conducted for assumed transition/transversion rate ratios (k) of 2 and 4. Support for nodes on the shortest tree and estimates of divergence time were derived using 1,000 bootstrap pseudoreplicates. The relative-rate test was performed to test the molecular clock hypothesis with MEGA 2 using Tajimas algorithm for clock hypotheses. In the final analysis, the phylogenetic trees were rooted using a representative species from each of the families, Dicroglossidae, Nyctibatrachidae, and Ranidae (Table 3).
 Back
Back Next
Next